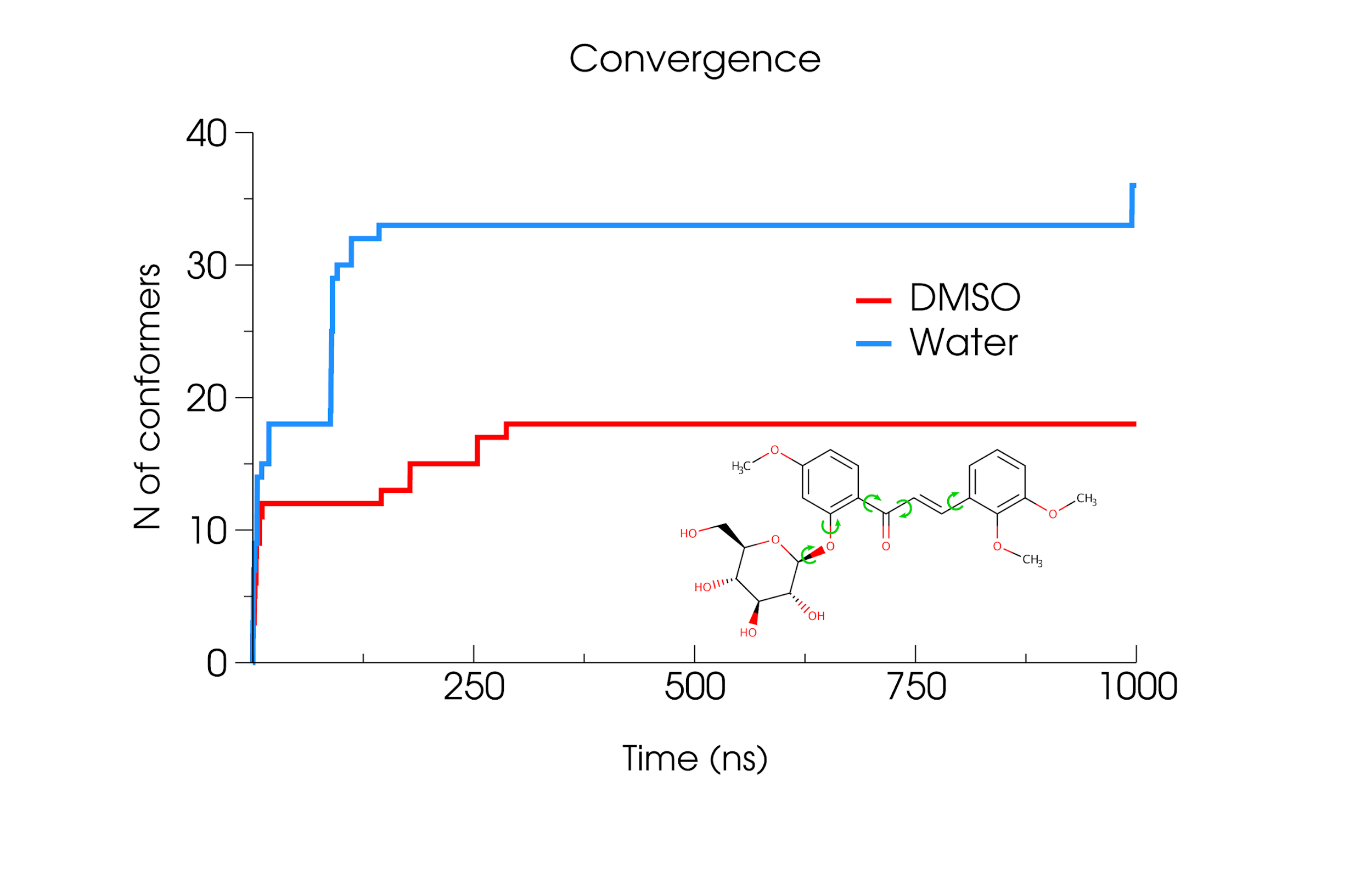
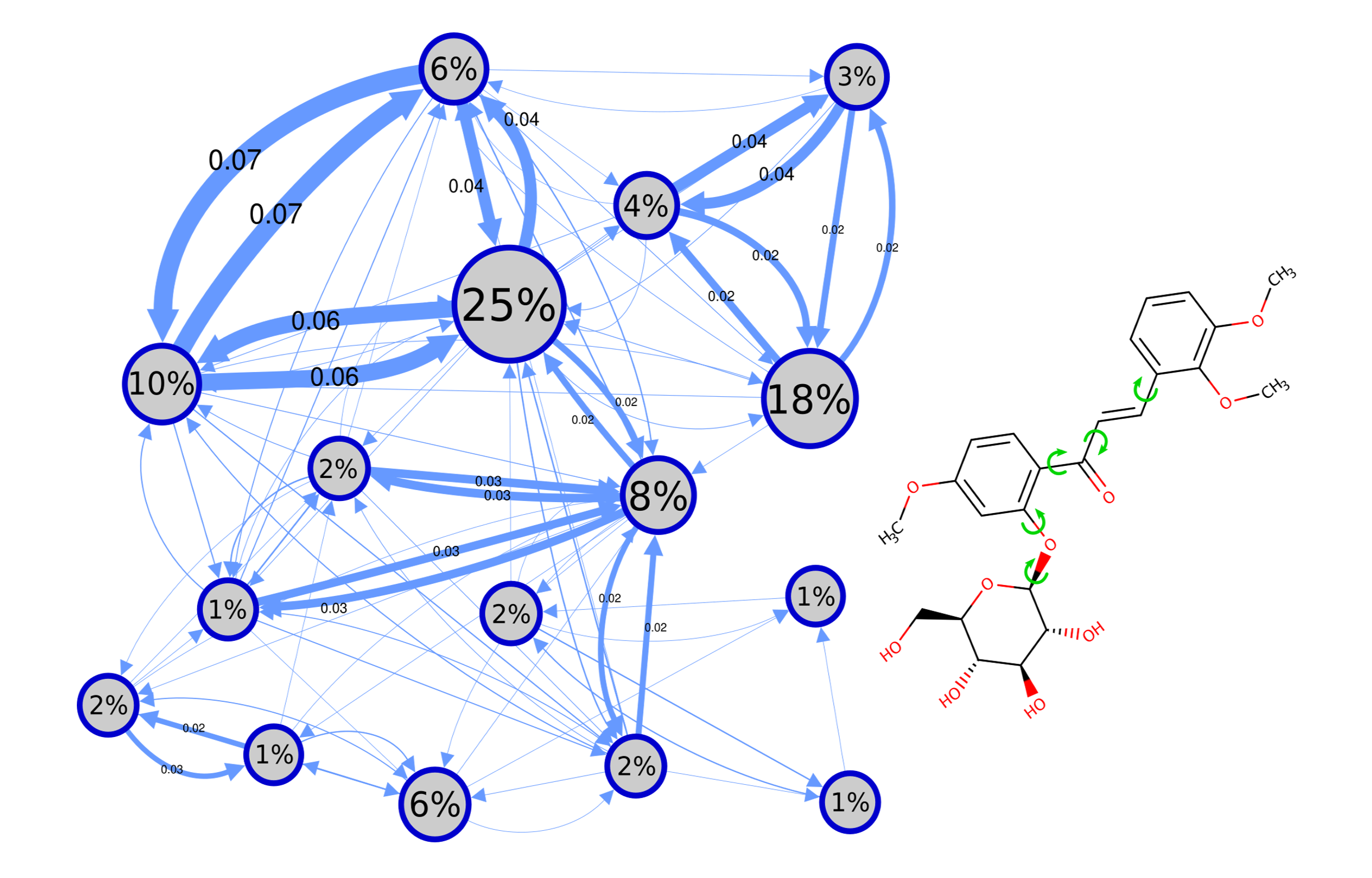
Conformational generation is a recurrent challenge in early
phases of drug design, mostly due to the task of making
sense between the number of conformers generated and
their relevance for biological purposes.
In this sense, ConfID, a Python-based
computational tool, was designed to identify and
characterize conformational populations of drug-like
molecules sampled through molecular dynamics simulations.
By using molecular dynamics (MD) simulations (and assuming
accurate parameters are used), ConfID can
identify all conformational populations sampled in the
presence of solvent and quantify their relative abundance,
while harnessing the benefits of MD and calculating
time-dependent properties of each conformational population
identified.

To contact us, please drop an email to bigrisci@inf.ufrgs.br and mdpoleto@vt.edu
What is ConfID?
It is a Python-based computational tool designed
to identify and characterize conformational populations
of small molecules sampled through molecular dynamics
simulations.
ConfID was developed by:
Bruno I. Grisci
- PhD
student (Institute of Informatics - UFRGS)
Marcelo D.
Polêto -
Postdoctoral Associate (Biochemistry Department - VirginiaTech)
Marcio Dorn -
Associate
Professor (Institute of Informatics - UFRGS)
Hugo Verli -
Associate
Professor (Center of Biotechnology - UFRGS)
To which problem ConfID was designed for?
Genetic algorithms and knowledge-based approaches have
been employed to study molecular flexibility. However,
these methods are usually based on crystallographic
information, and their calculations are made in vacuum
or with implicit solvent and do not take into account
the influence of explicit solvent molecules on
conformational preferences.
By using structural information gathered from MD
simulations (and assuming accurate parameters are
used), ConfID can identify all conformational populations
of drug-like molecules sampled in the presence of solvent
and quantify their relative abundance, while harnessing
the benefits of MD and calculating time-dependent properties
of each conformational population.

Tutorials
Frequently Asked Questions
Do you have any questions? Take a look at our FAQ
Publications
There are some papers already using ConfID! These are some:
- Feng, X., Li, F., Ding, M., Zhang, R., and Shi, T. Molecular Dynamic Simulation: Conformational Properties of Single-stranded Curdlan in Aqueous Solution, Carbohydrate Polymers, 2020
- Pablo R. Arantes, Conrado Pedebos, Marcelo D. Polêto, Laércio Pol-Fachin, and Hugo Verli. The Lazy Life of Lipid-Linked Oligosaccharides in All Life Domains, Journal of Chemical Information and Modeling 2020 60 (2), 631-643
- Pablo R. Arantes, Marcelo D. Polêto, Elisa B. O. John, Conrado Pedebos, Bruno I. Grisci, Marcio Dorn, and Hugo Verli. Development of GROMOS-Compatible Parameter Set for Simulations of Chalcones and Flavonoids, The Journal of Physical Chemistry B 2019 123 (5), 994-1008
- Roberta Tesch, Christian Becker, Matthias P. Müller, Michael E. Beck, Lena Quambusch, Matthäus Getlik, Jonas Lategahn, Niklas Uhlenbrock, Fanny N. Costa, Marcelo D. Polêto, Pedro S.M. Pinheiro, Daniel A. Rodrigues, Carlos M.R. Sant'Anna, Fabio F. Ferreira, Hugo Verli, Carlos A.M. Fraga, Daniel Rauh. An Unusual Intramolecular Halogen Bond Guides Conformational Selection, Angew. Chem. Int. Ed. 2018, 57, 9970
Citing ConfID
If you use ConfID in a scientific publication, we would appreciate citations to the following paper:
-
Marcelo D. Polêto, Bruno I. Grisci, Marcio Dorn, Hugo Verli.
ConfID: an analytical method for conformational characterization
of small molecules using molecular dynamics trajectories,
Bioinformatics. 2020
BibTeX@article{10.1093/bioinformatics/btaa130, author = {Polêto, M D and Grisci, B I and Dorn, M and Verli, H}, title = "ConfID: an analytical method for conformational characterization of small molecules using molecular dynamics trajectories", journal = {Bioinformatics}, year = {2020}, month = {02}, issn = {1367-4803}, doi = {10.1093/bioinformatics/btaa130}, url = {https://doi.org/10.1093/bioinformatics/btaa130}, note = {btaa130}, eprint = {https://academic.oup.com/bioinformatics/advance-article-pdf/doi/10.1093/bioinformatics/btaa130/32677172/btaa130.pdf}, }
Gallery